Important Facts For Prelims
Pompe Disease
- 16 Dec 2023
- 2 min read
Why in News?
India’s first Pompe disease patient, passed away at the age of 24 years after battling the disease in a semi-comatose state.
- A semi-comatose state is characterized by partial coma, manifesting as disorientation and stupor without reaching a complete coma. Individuals in a semi-comatose state may exhibit responsiveness to stimuli, such as groaning and mumbling.
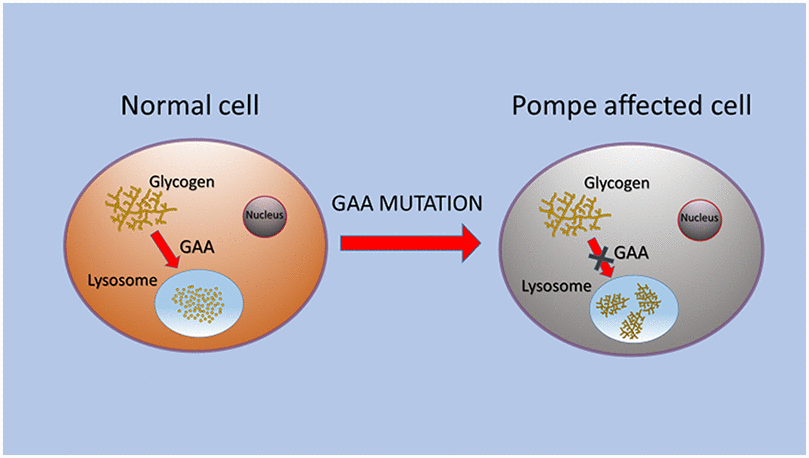
What is Pompe Disease?
- About:
- Pompe Disease (also known as Glycogen Storage Disease Type II) is characterized by the buildup of glycogen in the lysosomes of the body’s cells.
- This disease is a rare genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is crucial for breaking down glycogen into glucose within the lysosomes of cells.
- Lysosomes are membrane-enclosed organelles that contain an array of enzymes capable of breaking down all types of biological polymers—proteins, nucleic acids, carbohydrates, and lipids.
- Its prevalence estimates range from 1 in 40,000 to 1 in 300,000 births.
- Symptoms:
- Muscle weakness, Motor skill delay, Degenerative impact on bones, Respiratory complications, Cardiac involvement, Implications for daily living.
- Diagnosis:
- Enzyme assays are conducted to measure the activity of GAA, the deficient enzyme.
- Genetic testing identifies mutations in the responsible GAA gene. Genetic analysis confirms the presence of specific mutations associated with Pompe Disease.
- Treatment:
- Although there is presently no cure for Pompe disease, there are treatment alternatives accessible to address symptoms and enhance the patient's quality of life.
- Enzyme Replacement Therapy (ERT) is a common treatment method that entails infusing the deficient enzyme to mitigate glycogen accumulation.



